Rett syndrome pedigree: MECP2 inheritance, female bias and paternal de novo origin
A clinical guide to reading and counselling from a Rett syndrome pedigree — MECP2-driven X-linked dominance, the strong female bias, the paternal de novo mechanism, and the unusual combination of high penetrance with very low empirical recurrence risk.
Short version. Rett syndrome (ICD-10 F84.2; OMIM 312750) is an X-linked dominant neurodevelopmental disorder caused by pathogenic variants in MECP2 at Xq28. It is overwhelmingly a disease of females: hemizygous males with classical MECP2 variants are non-viable or severely affected neonatally. Penetrance in heterozygous females is approximately 99%, the de novo rate is approximately 99%, and germline mosaicism rate is approximately 0.5% with a paternal sex bias. The typical pedigree is an isolated affected female proband with unaffected parents and unaffected siblings. Recurrence risk to future daughters is very close to zero in practice because the variant overwhelmingly arises on the paternal X in an individual spermatogenic event, and recurrent transmission requires the rare situation of parental germline mosaicism.
Rett syndrome: clinical overview
Rett syndrome is a pervasive developmental disorder that classically presents in previously healthy girls between 6 and 18 months of age. Development is normal through the first half-year of life; a plateau period is followed by regression, with loss of acquired purposeful hand use, loss of spoken language, and appearance of stereotypic midline hand movements (hand wringing, washing, clapping, or mouthing). Gait disturbance ranges from ataxia to non-ambulation. Autonomic features are prominent: breath-holding, hyperventilation, breath-stacking, and air-swallowing. Seizures emerge in the majority of cases, typically in the preschool and school-age years, and can be multi-morphic. Growth deceleration, acquired microcephaly, kyphoscoliosis, bruxism, and prolonged QTc are characteristic.
The long-term phenotype includes severe intellectual disability, non-verbal communication, variable ambulation, and a fragile medical trajectory with increased risk of aspiration, respiratory compromise, cardiac conduction abnormalities, and sudden unexplained death. Life expectancy has improved markedly with modern multidisciplinary care. Trofinetide, an analogue of the N-terminal tripeptide of IGF-1, is the first approved disease-modifying therapy; supportive management and targeted symptomatic therapy for seizures, dystonia, and autonomic features remain the mainstay.
Atypical Rett variants exist. The preserved-speech (Zappella) variant retains some expressive language; the early-onset seizure variant historically overlapped with Rett but is now more commonly classified as CDKL5-related encephalopathy; the congenital variant is usually FOXG1-related. The pedigree considerations and recurrence parameters for CDKL5 and FOXG1 phenocopies are distinct from classical MECP2-related Rett syndrome.
MECP2 X-linked dominance and the female bias
MECP2 encodes methyl-CpG-binding protein 2, a transcriptional regulator highly expressed in neurons and crucial for neuronal maturation and synaptic function. Classical loss-of-function variants are incompatible with typical neurodevelopment. Because MECP2 sits on the X chromosome, male and female inheritance dynamics are asymmetric.
In a hemizygous male with a classical MECP2 loss-of-function variant, every neuron lacks functional MECP2; the outcome is typically pregnancy loss or severe neonatal encephalopathy. Surviving males with MECP2 variants generally fall into one of three categories: those with 47,XXY karyotype (who have a second X and express MECP2 mosaically like females); those with somatic mosaicism for a de novo post-zygotic MECP2 variant; or those with non-classical hypomorphic MECP2 alleles producing the distinct MECP2-duplication-syndrome-adjacent phenotype. Classical Rett syndrome in a phenotypically typical 46,XY male is exceptional.
In a heterozygous female with a classical MECP2 variant, random X-inactivation produces a mosaic of MECP2-expressing and MECP2-silent neurons. Each cell expresses either the maternal or paternal X chromosome but not both, and the inactivation pattern is typically fixed early in development. The resulting 50:50 mosaic produces the characteristic phenotype; skewing of X-inactivation towards the wild-type allele can ameliorate severity, and skewing towards the variant allele can worsen it.
X-linked dominance with female predominance is the rule; a conventional X-linked dominant pedigree with multiple affected females across generations, as seen in disorders such as incontinentia pigmenti, is not the usual Rett syndrome picture because reproduction in classical Rett is rare.
The paternal de novo mechanism
Approximately 99% of classical Rett syndrome cases are de novo. The variant arose as a new mutation and was not inherited from an affected parent. An important mechanistic detail: the overwhelming majority of de novo MECP2 variants arise on the paternal X chromosome. The most common variants are C-to-T transitions at methylated CpG dinucleotides (T158M, R168X, R255X, R270X, R294X, R306C) — a mutational signature associated with the higher mutation rate in spermatogenesis and the characteristic mutability of methylated CpGs in male germline.
The practical counselling consequence is asymmetric. A father's sperm are produced continually from spermatogonial stem cells; an individual de novo MECP2 variant in a single sperm that produced the proband does not carry forward into the father's subsequent sperm unless the mutation arose early in spermatogonial development (in which case the father would be germline-mosaic, a rare event at approximately 0.5% prior). A mother's oocytes are fixed in number at birth; if the variant arose in the mother's germline, it would be present as a mosaic in her oocyte population, but maternal de novo events are the minority mechanism in MECP2-related Rett syndrome, and maternal germline mosaicism is correspondingly rare.
For a contrasting X-linked recessive example in which maternal germline mosaicism dominates the residual recurrence risk, see our X-linked recessive calculator guide. For the general autosomal dominant arithmetic, see our autosomal dominant calculator.
A pedigree example
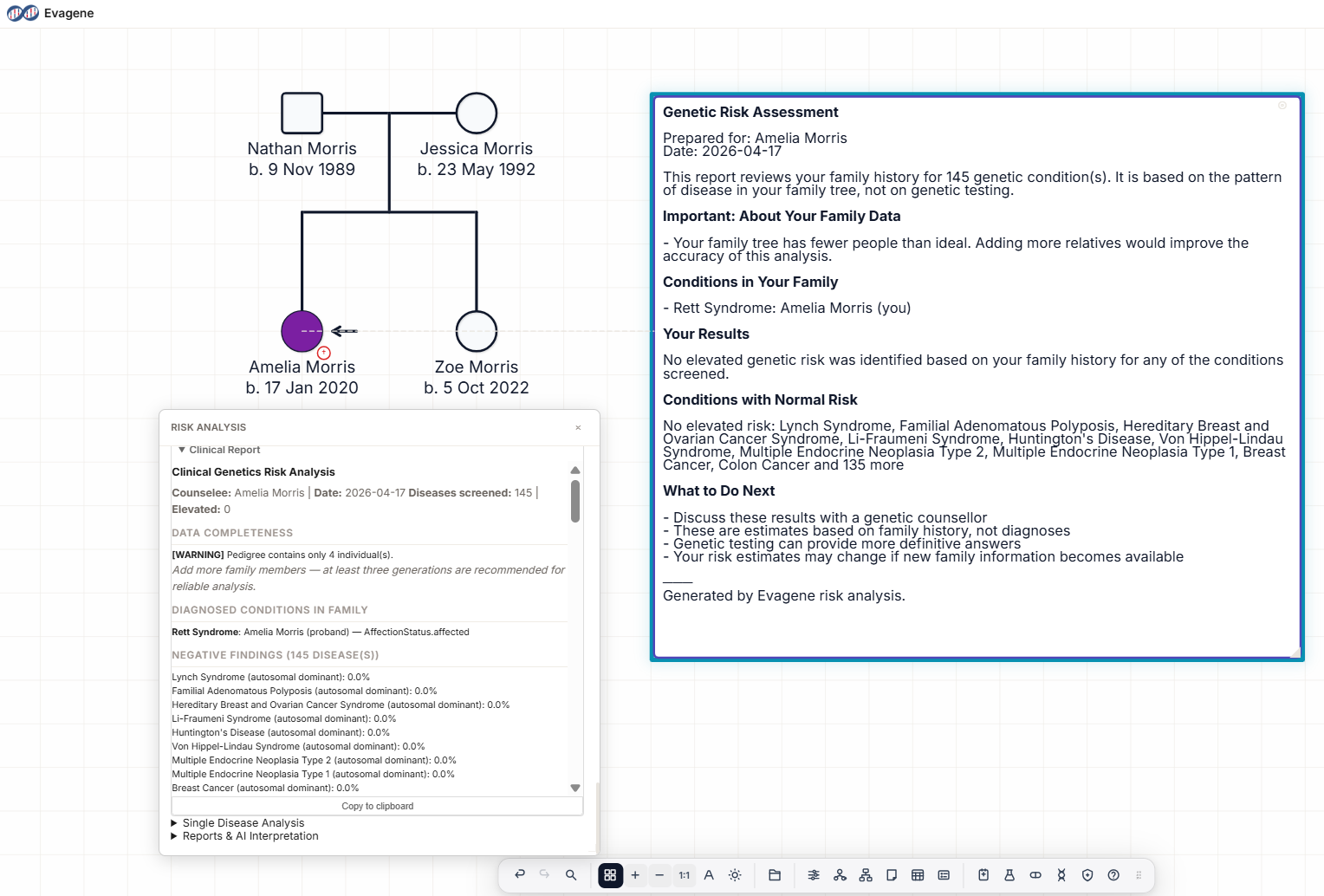
The pedigree above shows the typical Rett syndrome family. An unaffected father (unshaded square) and an unaffected mother (unshaded circle) have three children: the proband is a filled circle with the arrow identifying her as the proband, flanked by unshaded square and circle siblings. Disease annotation under the proband reads "Rett syndrome (F84.2); MECP2 c.[insert] p.[insert]; de novo; parental testing negative".
Read the pedigree in two steps. First, the proband's phenotype and molecular confirmation: classical Rett syndrome clinical criteria met, an MECP2 pathogenic variant identified, and both parents negative for the variant on blood testing. Second, the pedigree structure as evidence for or against parental germline mosaicism: a single affected child with two unaffected siblings and two parents with negative blood testing is consistent with de novo paternal origin; it does not force the mosaicism hypothesis. Where available, sperm sequencing in the father can directly estimate the germline allele fraction and tighten the recurrence estimate.
Recurrence risk: high penetrance, low empirical recurrence
Rett syndrome is an unusual combination of high penetrance (~99% in heterozygous females) with very low empirical recurrence risk. The gap is explained by the mechanism: variants arise de novo in individual germ cells, and those germ cells are not representative of the parental germline compartment unless mosaicism is present at an early developmental stage.
Counselling positions, in outline:
- Isolated proband, both parents blood-negative, no other affected sibs: recurrence risk for future daughters is very low — commonly counselled at under 1% — with the residual risk driven by unobserved parental germline mosaicism, predominantly paternal.
- Measurable paternal sperm VAF: recurrence risk scales with sperm VAF and half transmission probability. A measurable positive sperm fraction substantially elevates the recurrence estimate; a negative sperm result at adequate sensitivity further lowers it.
- Affected female proband who reaches reproductive age (rare): 50% transmission per pregnancy to either sex; affected daughters expected to develop Rett syndrome, affected sons typically non-viable.
- Two affected female siblings with parents blood-negative: posterior probability of parental germline mosaicism approaches certainty and recurrence is driven by the germline fraction; by far the most likely parent is the father given the paternal bias.
A practical note on counselling tone: families whose child has just received a Rett syndrome diagnosis are often absorbing grief alongside the technical genetics. The fact that recurrence risk is very low is usually reassuring, but should be delivered with the honest caveat that "very low" is not zero and that the residual risk is quantifiable if the family wishes — through paternal sperm sequencing and through prenatal or preimplantation testing in subsequent pregnancies.
How Evagene supports Rett syndrome pedigrees
Rett syndrome is a first-class catalogue entry in Evagene. The record carries ICD-10 F84.2, OMIM 312750, X-linked dominant inheritance, penetrance 0.99 in females, a de novo rate of 0.99, and a germline mosaicism rate of 0.005 with paternal sex bias and female-specific expression. Annotating the proband with Rett syndrome automatically exposes these condition-specific parameters to the downstream analysis surface, including the nuanced paternal-biased germline mosaicism that distinguishes Rett from most other X-linked disorders.
The germline mosaicism posterior accepts pedigree structure and optional blood and sperm VAF for each parent, and applies the paternal bias encoded in the catalogue parameters. For an isolated proband with blood-negative parents, the posterior for each parent is low; for two affected siblings with the same MECP2 variant, the posterior concentrates strongly on the father with the bias; for a measurable paternal sperm VAF, the posterior becomes a direct function of that fraction. The output is a transparent calculation rather than an empirical rule-of-thumb, which lets a counsellor explain the recurrence figure with its assumptions.
AI-assisted clinical interpretation using bring-your-own-key Anthropic Claude or OpenAI GPT models generates a structured report covering MECP2 inheritance, the paternal de novo mechanism, the mosaicism posterior with its assumptions, the cascade testing plan (typically minimal given the low recurrence), available reproductive options (prenatal and preimplantation testing), and the disease-specific management summary including trofinetide eligibility. The rare disease pedigree software overview sets out the broader rare-disease analysis pipeline; the companion Dravet syndrome pedigree page is a useful contrast for autosomal dominant de novo disorders.
Frequently asked questions
What is the inheritance pattern of Rett syndrome?
X-linked dominant with a strong female bias. Classical MECP2 loss-of-function variants are male-lethal in utero or produce severe neonatal encephalopathy. Female penetrance is approximately 99%.
Why is Rett syndrome almost exclusively female?
MECP2 is essential in neurons. Hemizygous males have no wild-type copy; heterozygous females survive because random X-inactivation leaves a mosaic of MECP2-expressing cells.
What is the typical Rett pedigree?
An isolated affected female proband with unaffected parents and siblings. Multiplex Rett families are rare.
What is the recurrence risk after an isolated case?
Very low, typically counselled below 1% with parents blood-negative. Residual risk is driven by paternal germline mosaicism, which is rare.
Which MECP2 variants cause Rett?
Eight recurrent variants account for most cases: T158M, R168X, R255X, R270X, R294X, R306C, and C-terminal frameshifts. Other loss-of-function variants also cause the phenotype.
What is the difference between classical and atypical Rett?
Classical Rett meets defined clinical criteria and is usually MECP2-related. Atypical variants include the preserved-speech Zappella variant, and CDKL5- and FOXG1-related phenocopies, which have distinct pedigree considerations.
How does Evagene handle Rett syndrome?
Rett is a first-class catalogue entry with OMIM, ICD-10, inheritance, penetrance, de novo rate, and paternally-biased germline mosaicism parameters. X-linked dominant analysis and the joint-parent mosaicism posterior with sperm VAF input run directly on the pedigree.