Tuberous sclerosis pedigree: TSC1/TSC2 inheritance, mosaicism and reduced penetrance
A clinical guide to reading and counselling from a tuberous sclerosis complex pedigree — TSC1 and TSC2 autosomal dominance, the striking intrafamilial variable expressivity, and the posterior distribution that weighs mosaicism against inherited reduced penetrance.
Short version. Tuberous sclerosis complex (TSC; ICD-10 Q85.1; OMIM 191100 for TSC1 and 613254 for TSC2) is an autosomal dominant multisystem disorder caused by loss-of-function variants in TSC1 on 9q34 or TSC2 on 16p13.3. Penetrance is approximately 95% but with substantial intrafamilial variable expressivity — two members of the same family with the identical variant can have very different disease severity. Approximately 65% to 70% of cases are de novo; germline mosaicism contributes an additional ~1% to 2%. The instructive counselling scenario is three affected children born to two phenotypically unaffected parents with negative blood testing: the posterior distribution between parental germline mosaicism (~48%) and an inherited reduced-penetrance allele in a mildly or subclinically affected parent (~52%) is approximately balanced, with a computed recurrence risk for the next pregnancy in the region of 35%. This case requires careful parental phenotyping, sensitive molecular testing, and explicit handling of both hypotheses in the counselling discussion.
Tuberous sclerosis: clinical overview
TSC is a genetic multisystem disorder characterised by benign tumours — hamartomas — in the brain, skin, kidneys, heart, lungs, and eyes. Birth prevalence is approximately 1 in 6,000 to 1 in 10,000.
- Central nervous system: cortical and subcortical tubers, subependymal nodules, and subependymal giant cell astrocytomas (SEGAs). Epilepsy occurs in approximately 80% to 90% of individuals and often presents in infancy as infantile spasms. Intellectual disability and autism spectrum disorder are common. "TSC-associated neuropsychiatric disorders" (TAND) capture the broader cognitive and behavioural phenotype.
- Skin: hypomelanotic macules ("ash-leaf spots") visible with Wood's lamp from infancy, facial angiofibromas (previously called adenoma sebaceum), shagreen patches (lumbosacral), periungual fibromas, fibrous cephalic plaques, confetti skin lesions.
- Kidneys: angiomyolipomas, often multiple and bilateral, with risk of spontaneous haemorrhage; epithelial cysts; a modestly increased lifetime risk of renal cell carcinoma.
- Heart: cardiac rhabdomyomas are characteristic in infancy and often regress; fetal rhabdomyomas may be the presenting feature.
- Lungs: lymphangioleiomyomatosis (LAM) in adult women, with progressive cystic lung disease and risk of pneumothorax.
- Eyes: retinal hamartomas and retinal pigmentary lesions.
Management centres on vigilance for SEGA progression (serial brain MRI), surveillance of angiomyolipomas, seizure management, and TAND assessment. mTOR inhibitors (everolimus, sirolimus) are disease-modifying: they reduce SEGA volume, angiomyolipoma size, facial angiofibromas, and seizure burden, and slow LAM progression. Vigabatrin remains first-line for infantile spasms in TSC. The 2021 updated diagnostic criteria combine major and minor clinical features with genetic testing as an independent diagnostic route.
TSC1 and TSC2 genetics and variable expressivity
TSC1 encodes hamartin and TSC2 encodes tuberin; together they form the TSC1-TSC2 complex that inhibits the mTOR pathway. Loss of either protein releases mTOR signalling, producing the hamartomatous phenotype. TSC2 variants account for approximately two thirds of cases overall, and TSC2-related disease is on average more severe than TSC1-related disease.
The mutation spectrum includes nonsense, frameshift, splice-site, and large deletions affecting either gene; a specific subset, the contiguous deletion of TSC2 and the adjacent PKD1 gene, produces a combined TSC-polycystic kidney disease phenotype. No-mutation-identified (NMI) TSC accounts for approximately 10% of clinically diagnosed cases, often driven by deep intronic variants, low-level mosaicism undetected by standard sequencing, or non-coding regulatory variants.
The hallmark TSC genetics feature — and the one most relevant to pedigree counselling — is variable expressivity. Two family members with the same pathogenic variant can have markedly different phenotypes. One may have severe early-onset epilepsy, intellectual disability, and autism; another may have only a few hypomelanotic macules and be otherwise well. This variability is partly stochastic (the second-hit somatic-loss-of-heterozygosity events driving hamartoma formation occur at different times, locations, and numbers in different individuals) and partly the cumulative effect of modifier alleles.
Penetrance estimates for clinically identifiable TSC are in the region of 95% but this figure glosses over the clinical reality: a proportion of "penetrant" carriers have subclinical or previously unrecognised disease, which is why careful phenotyping of relatives changes the counselling picture.
The three-affected-children scenario
Consider a couple with three children, all diagnosed with TSC and all carrying the same pathogenic TSC2 variant. Both parents report no clinical features of TSC. Blood testing in both parents is negative for the variant with appropriate sensitivity. There is no TSC in the extended family. The question — what is the recurrence risk for a fourth pregnancy? — requires a careful posterior calculation.
Two hypotheses compete:
- Parental germline mosaicism. One parent carries the variant in a fraction of their germline but not in their somatic blood. The fraction is stable enough to produce three affected children by chance. Recurrence per future conception under this hypothesis depends on the germline fraction estimate.
- Inherited reduced penetrance or subclinical parental TSC. One parent carries the variant constitutionally but has a mild or subclinical phenotype, was never diagnosed, and the blood test is falsely negative or the variant is present at an atypical level. Recurrence per future conception under this hypothesis is ~50% × penetrance.
Given a TSC germline mosaicism prior of ~0.01-0.02 and the substantial variable expressivity of TSC, the posterior distribution after observing three affected children is approximately balanced: ~48% mosaicism, ~52% inherited. The recurrence risk for a subsequent pregnancy, computed as a weighted combination, lands around 35% — substantially higher than the de-novo posterior from a single affected case would have suggested, and materially different between the two explanations (~13% under the mosaicism-only scenario versus ~48% under the inherited-penetrance scenario, with the posterior weighting them).
A pedigree example
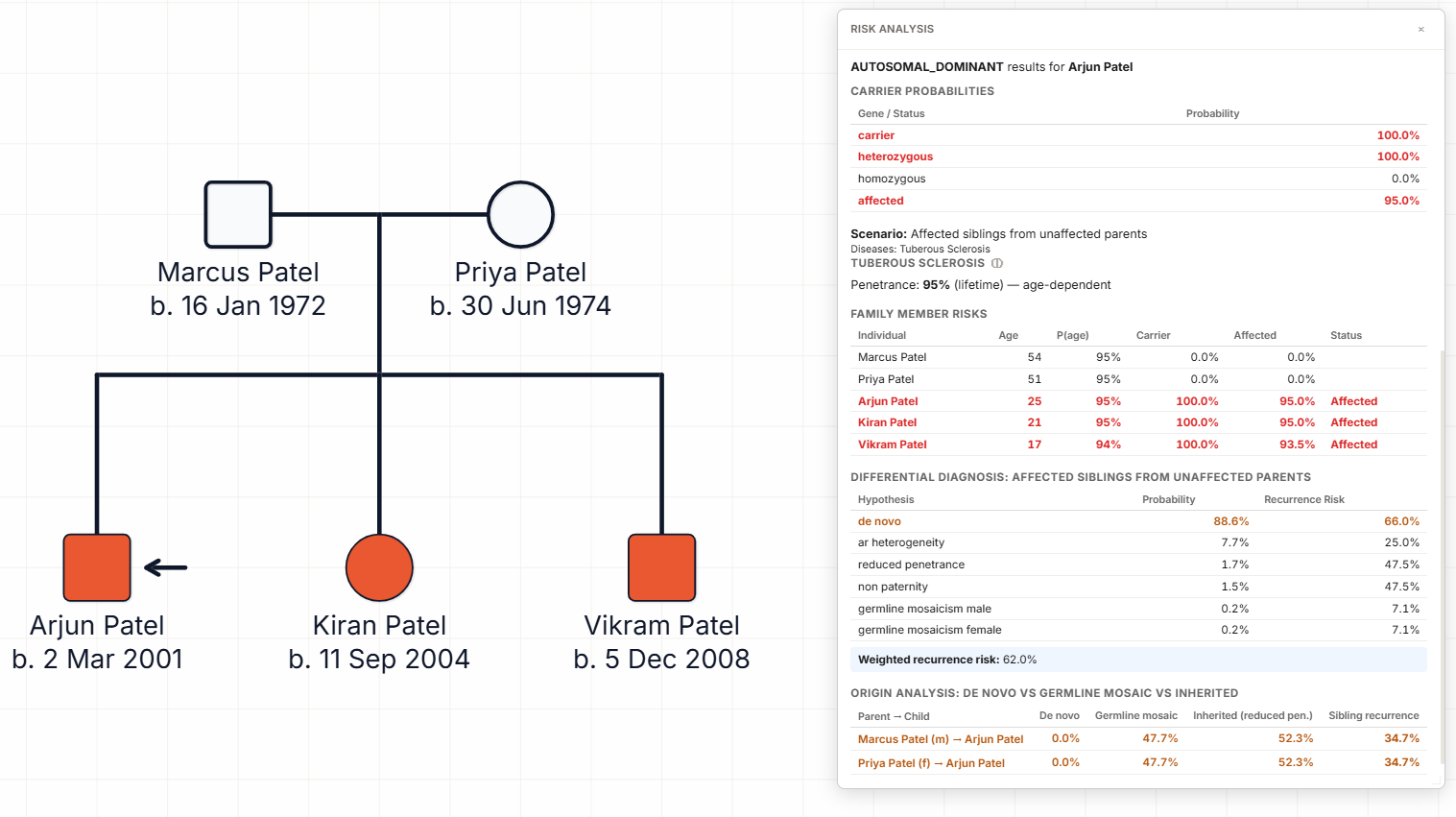
The pedigree above shows the three-affected-children scenario. Two phenotypically unaffected parents (unshaded square and circle) have three children, all filled symbols, all annotated with the same TSC2 pathogenic variant and TSC clinical features documented in the patient record. A fourth position in the sibship is drawn as a lozenge with a question mark, representing the ongoing pregnancy under discussion.
Reading the pedigree is a two-step process:
- Step 1: Rule out parental clinical TSC. Wood's lamp examination of both parents for hypomelanotic macules; renal ultrasound for angiomyolipomas or cysts; brain MRI for cortical tubers, subependymal nodules, or SEGAs; cardiac echo in infancy parents may have missed; fundoscopy for retinal hamartomas; review of medical history for unexplained seizures, renal haemorrhage, or pneumothorax. Even a single minor feature in either parent substantially changes the posterior in favour of inherited reduced penetrance.
- Step 2: Quantify the posterior. With both parents phenotypically negative and blood-negative at appropriate sensitivity, run the joint-parent germline mosaicism posterior incorporating the pedigree structure (three affected from three) and the TSC-specific priors. The output is a probability distribution between the two hypotheses and a recurrence estimate with its assumptions transparent.
Sperm sequencing in the father can further refine the estimate if it is positive (concentrating the posterior on paternal germline mosaicism and giving a direct germline fraction), though a negative sperm result does not rule out maternal mosaicism. Where available, tissue-specific sampling in either parent can also help — hypomelanotic macules on the parent, if present, can be biopsied for targeted sequencing to detect low-level somatic TSC variants.
Counselling implications
The practical counselling messages in the three-affected-children TSC scenario:
- Recurrence is materially high. A figure of around 35% is very different from the background prior, different from the single-affected-case de novo residual, and demanding of a full reproductive counselling conversation that includes prenatal diagnosis, preimplantation genetic testing, and the option of not proceeding with further pregnancies.
- The recurrence is uncertain. The posterior distribution is broad. The family should understand that the 35% figure is a weighted average over two hypotheses with different consequences, and that the range around it is clinically important.
- Detailed parental phenotyping changes the answer. Finding any TSC feature in a parent concentrates the posterior on inherited reduced penetrance and raises the recurrence estimate towards ~48%. Finding none, combined with negative deep sequencing, concentrates it on mosaicism and lowers the figure towards ~13%.
- Affected children's own reproductive counselling differs. Each affected child, as they reach reproductive age, has a 50% transmission risk to their own offspring regardless of how the variant entered the family, and their own children will have the same variable expressivity.
- Surveillance in affected children is independent of recurrence counselling. Each affected child needs standard TSC surveillance and management regardless of how the pedigree is interpreted.
For the autosomal dominant transmission arithmetic underlying these figures, see our autosomal dominant calculator. For the germline mosaicism posterior calculation, see our germline mosaicism calculator.
How Evagene supports tuberous sclerosis pedigrees
Tuberous sclerosis complex is a first-class catalogue entry in Evagene. The record carries ICD-10 Q85.1, OMIM 191100 for TSC1-related TSC and 613254 for TSC2-related TSC, autosomal dominant inheritance with annotation on variable expressivity, a penetrance parameter around 0.95, a de novo rate of ~0.65 to 0.7 combined across genes, and a germline mosaicism rate of ~0.01 to 0.02. Annotating the proband with TSC makes these parameters available to the analysis surface.
The joint-parent germline mosaicism posterior models the two competing hypotheses — parental germline mosaicism and inherited reduced penetrance — simultaneously. The output is a posterior probability for each, together with a recurrence estimate computed as the weighted combination. Input parameters can be refined by adding parental phenotypic findings, measured blood VAF, or measured sperm VAF, and the posterior updates accordingly in real time. The hereditary cardiac pedigree guide provides a related pattern for cardiac rhabdomyomas presenting antenatally.
AI-assisted clinical interpretation using bring-your-own-key Anthropic Claude or OpenAI GPT models generates a structured report covering the TSC diagnostic criteria satisfied, the molecular detail, the inheritance pattern, the posterior distribution between mosaicism and reduced penetrance, the parental assessment plan, the reproductive options, and the TSC surveillance schedule for each affected individual. For the broader rare-disease analysis workflow see our rare disease pedigree software overview.
Frequently asked questions
What is the inheritance pattern of TSC?
Autosomal dominant, caused by TSC1 or TSC2 variants. Penetrance approximately 95% with substantial intrafamilial variable expressivity. Around 65-70% of cases are de novo.
What is the three-affected-children scenario?
Three affected children from two blood-negative unaffected parents. Posterior distribution is roughly balanced between parental germline mosaicism (~48%) and inherited reduced penetrance (~52%); recurrence approximately 35%.
What are the clinical features of TSC?
Cortical tubers, SEGA, epilepsy, intellectual disability, autism spectrum, facial angiofibromas, hypomelanotic macules, shagreen patches, cardiac rhabdomyomas, renal angiomyolipomas, LAM (adult women), and retinal hamartomas.
How does Evagene distinguish mosaicism from reduced penetrance?
The joint-parent posterior models both hypotheses simultaneously and returns a probability for each, with a weighted recurrence estimate. Parental phenotyping and molecular findings update the posterior.
What is the recurrence risk in this scenario?
Approximately 35% for the next pregnancy, weighted across the mosaicism and reduced-penetrance hypotheses. Detailed parental assessment can shift the figure materially in either direction.
Can an affected parent be subclinical in TSC?
Yes — this is characteristic of TSC. Careful Wood's lamp, renal ultrasound, brain MRI, fundoscopy, and medical history review can identify otherwise unrecognised parental involvement.
How does Evagene handle TSC?
TSC is a first-class catalogue entry with OMIM, ICD-10, inheritance, penetrance, de novo rate, variable expressivity notation, and mosaicism parameters. The joint-parent posterior, AI interpretation, and report generation all run directly on the pedigree.